1PTE ÁOK, Sebészeti Oktató és Kutató Intézet, Pécs
2Zala Megyei Kórház, Szívsebészeti Osztály, Zalaegerszeg
3PTE ÁOK, Anaesthesiológia és Intenzív Therápiás Intézet, Pécs
4PTE ÁOK, Sebészeti Oktató és Kutató Intézet, Pécs
5PTE ÁOK, Sebészeti Oktató és Kutató Intézet, Pécs
6PTE ÁOK, Sebészeti Oktató és Kutató Intézet, Pécs
7PTE ÁOK, Sebészeti Oktató és Kutató Intézet, Pécs
8PTE ÁOK, Sebészeti Oktató és Kutató Intézet, Pécs
9Faculty of Medicine, Imperial College London, The Royal Brompton and Harefield NHS Foundation Trust, Harefield, United Kingdom
A glutation-S-transzferáz gátlásának hatása oxidatív stresszel károsított szívizomsejtekre
2011-12-14Az oxidatív stressz és az iszkémiás károsodás fontos szerepet játszik számos kardiovaszkuláris betegség patogenezisében, illetve gyakori velejárója lehet a különböző klinikai, többek között sebészeti beavatkozásoknak is. A glutation-S-transzferáz (GST) jól ismert antioxidáns enzim, amely az oxidatív stressz során keletkező anyagcsere-termékek nagy kapacitású inaktivációjában és ezáltal a sejtkárosodás kivédésében játszik szerepet. A GST aktivitásának megváltozása befolyásolhat számos jelátviteli mechanizmust, így például kulcsfontosságú szerepe van a mitogén aktivált protein-kináz (MAPK) jelátviteli utak szabályozásában és ezáltal a stresszre adott sejtválaszban. Kísérleteink fő célja etakrinsavval létrehozott GST-gátlás vizsgálata oxidatív stressznek, illetve szimulált iszkémia-reperfúziónak kitett szívizom-sejttenyészeten.
Vizsgálatainkat újszülött patkányból izolált primer szívizomsejteken végeztük, amelyet az alkalmazott kezelések szerint 6 csoportra osztottunk, az etakrinsav, H2O2 és szimulált iszkémia-reperfúziós kezelés, illetve ezek kombinációja szerint. A sejtek életképességét MTT-próbával, az apoptotikus sejtek mennyiségét annexin V/propidium-jodid kettős festést követő flow-citometriás méréssel határoztuk meg. A GST-gátlás jelátviteli útvonalakra való hatását a proapoptotikus JNK és p38 mitogén aktivált protein-kinázok, és az antiapoptotikus ERK/p42-p44 MAPK és protein-kináz B(PKB)/Akt aktivitásának flow-citometriás mérésével vizsgáltuk.
Az etakrinsav által létrehozott GST-gátlás a sejtek életképességének jelentős csökkenéséhez vezetett és növelte az apoptotikus sejtek számát. Az etakrinsav által létrehozott GST-gátlás már önmagában fokozta a proapoptotikus jelátviteli utak (JNK, p38), valamint az antiapoptotikus ERK/p42-p44 aktivációját, míg a szintén antiapoptotikus PKB/Akt aktivitását jelentősen csökkentette. Amennyiben a sejteket etakrinsavas kezelés mellett egyidejű oxidatív stressznek is kitettük, a sejtkárosító szignálok fokozottabb aktivációját figyeltük meg. Eredményeink igazolják a GST kulcsfontosságú szerepét a szívizomsejtek túlélésében és a MAP-kináz jelátviteli utak szabályozásában oxidatív stressz és szimulált iszkémia-reperfúziós károsodás során.
Az oxidatív stressz kialakulása bármilyen szerv iszkémiás-reperfúziós károsodását követően, a sejtekben fellépő apoptotikus, nekrotikus elváltozások kialakulásához vezethet. Ezen állapotban a rendkívül agresszív reaktív oxigén-intermedierek (ROI) képződése meghaladja az endogén antioxidáns védelem kapacitását, s így valamennyi biomolekula (zsírok, fehérjék, szénhidrátok) oxidatív károsodását idézik elő (1–5).
Az utóbbi évek irodalmi adatai felhívták a figyelmet arra, hogy a sejtszintű elváltozások mélyebb megismerése számos előnyt jelenthet a klinikai gyakorlatban is. Olyan esetekben, amikor a tervezett műtétek során (ér- és szívsebészet, szervtranszplantáció) elkerülhetetlen a rövidebb-hosszabb időre kialakuló iszkémia, majd a keringés helyreállítását követő reperfúzió, lehetőség van előre felkészíteni a szervezetet az endogén antioxidáns védelem indukálása révén (6–8). Ezen a területen ma „gold standardnak” tekinthető pre- és posztkondicionálás már a klinikai gyakorlatban is használt (9).
A reaktív gyökök elleni védelemben a máj által aminosavakból szintetizált glutationnak kiemelten fontos szerepet tulajdonítunk, hisz részben az antioxidáns glutation-peroxidáz szubsztrátjaként szerepel, miközben oxidált formává, GSSG-vé alakul át. A GSH/GSSG arány a plazmában jól mérhető és kóros állapotokban tükrözi a szervezetben kialakuló oxidatív folyamatok intenzitását (10). Ugyancsak a redukált glutationt használja szubsztrátként a glutation-S-transzferáz (GST) enzim az elektrofil vegyületek és toxikus anyagok közömbösítése során. Az utóbbi években mutatták ki a GST-jelátvitelben, génexpresszióban, apoptózisban, fehérje glutationilációban, a nitrogén-oxid metabolizmusban, valamint a gyulladásban betöltött fontos szerepét (11–13).
Munkánk során a GST-enzim szerepét vizsgáltuk oxidatív stressznek, illetve iszkémia-reperfúziónak kitett szívizomsejteken. Kísérleteink célja elsősorban az volt, hogy meghatározzuk a GST-gátlás hatását a szívizomsejtek viabilitására, az apoptózis mértékére, valamint a mitogén aktivált protein (MAP) kináz jelátviteli utak esetleges befolyásolására.
Anyagok és módszerek
Sejtkultúra
Vizsgálatainkat újszülött patkányból izolált primer szívizomsejt-tenyészeten végeztük korábban már leírt protokoll alapján (14, 15). 2-4 napos Wistar-patkányokból kamrai szívizomsejteket nyertünk kollagenáz (GibcoTM Collagenase Type II, Invitrogen Corp., Carlsbad, CA, USA) segítségével. Az izolált sejteket I-es típusú kollagénnel (GibcoTM Collagenase Type II, Invitrogen Corp., Carlsbad, CA, USA) fedett tenyésztőedényekbe helyeztük 200.000/cm2 sűrűségben. A sejteket 10% fetal bovine szérummal (FBS, Gibco, USA) kiegészített DMEM/F12 (Sigma–Aldrich, USA) médiumban inkubáltuk. A következő napon, amikor a sejtek már szilárdan kitapadtak a tenyésztőedény aljához, a tápoldatot komplett szérummentes médiumra (CSFM: 2,5 mg/ml BSA, 1 µM inzulin, 5,64 µg/ml transzferrin, 32 nM szelénium, 2,8 mM Na-piruvát, 0,1-1 nM T3, 100 IU/ml penicillin, 0,1 mg/ml streptomycin, 200 mM L-Glutamin, DMEM/F12) cseréltük. A kísérletet 24 órás inkubációt követően kezdtük, és a médiumot 24 óránként cseréltük. A szívizomsejteket random módon 6 csoportba osztottuk az alkalmazott kezelések szerint.
A nem kezelt sejteket a kísérlet teljes ideje alatt komplett szérummentes médiumban (CSFM) tartottuk, amelyek pozitív kontrollként szolgáltak (I. csoport). A II. csoportban a médiumhoz etakrinsavat (150 µM EA, Sigma) adtunk a GST-enzim gátlására. A III. csoportban 1 mM-os hidrogén-peroxidot (H2O2) alkalmaztunk az oxidatív stressz indukálására. A IV. csoportban együtt alkalmaztuk a 150 µM etakrinsavas-gátlást és az 1 mM-os H2O2-kezelést. Az V. csoportban megfelelő médiumok cseréjével, a szívizomsejteket szimulált iszkémia-reperfúziónak (I/R) tettük ki, míg a VI. csoportban 150 µM etakrinsavat is hozzáadtunk a kezelt sejtekhez.
Azoknál a csoportoknál, ahol szimulált I/R-t alkalmaztunk, a sejteket 1,5 órás iszkémiának vetettük alá, amihez korábban leírt iszkémiás puffert használtunk (16), majd ezt követte 2,5 óra reperfúzió, ami az iszkémiás puffer CSFM-re cserélésével történt. A VI-os csoportnál, ahol a sejtek I/R- és EA-kezelést is kaptak, mind az iszkémiás mind a reperfúziós médium tartalmazta az etakrinsavat 150 µM-os koncentrációban.
Az etakrinsav koncentrációját, megelőző kísérleteink alapján választottuk 150 µM-nak, a kezelési időt pedig 4 órának.
A sejtek életképességének vizsgálatára a kezelés befejezése után azonnal sor került. Az apoptotikus jelátviteli markerek meghatározása szintén a kezelés végeztével kezdődött és a permeabilizációig tartott, majd a mintákat így tároltuk –20 °C-on a további feldolgozásig.
Sejtéletképesség vizsgálata
A sejtek életképességét MTT mérési módszerrel határoztuk meg (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide, Sigma). A módszer alapja, hogy az élő sejtek működőképes mitokondriumai az MTT-t redukálja kék formazán festékké. Ennek abszorbanciáját ELISA-olvasóval mértük meg (Sirio microplate reader, Seac Corp. Florence, Italy), 570 nm-en és a kapott értékeket (arbitrary units; AU) ábrázoltuk. Az eredményeket százalékban fejeztük ki a kontrollértékekhez viszonyítva.
A sejtek apoptózis vizsgálata
Az élő és az apoptotikus sejtek arányát fluoreszcein izoticianát (FITC) jelölt annexin V (BD Biosciences, Pharmingen, USA) és propidium-jodid (BD Biosciences, Pharmingen, USA) kettős festést követően flow-citometriával határoztuk meg korábban leírtak alapján (17). A mintákat BD FacsCalibur flow-citométerrel mértük (BD Biosciences, USA) és cellquest szoftverrel analizáltuk. A sejteket minden csoportban az összes festődött sejt százalékának arányában adtuk meg.
Jelátviteli markerek vizsgálata
A GST-gátlás jelátviteli útvonalakra való hatását a proapoptotikus mitogén aktivált protein-kinázok (MAPK), mint a Jun N-terminális kináz (JNK), p38, és az antiapoptotikus extracelluláris szignál által regulált kináz (ERK/p42-p44), valamint a protein-kináz-B (PKB/Akt) aktivitásának flow-citometriás mérésével vizsgáltuk. A minták analizálása az előbbiekben említett flow-citométerrel történt. A sejteket minden csoportban az összes festődött sejt százalékának arányában adtuk meg.
Statisztika
Az összes adatot az átlag±az átlag standard hibájaként (S.E.M) adtuk meg. A csoportok közötti különbségeket egyirányú ANOVA és Student t-teszttel értékeltük ki. A p-értékét szignifikánsnak tekintettük, ha kisebb volt, mint 0,05.
Eredmények
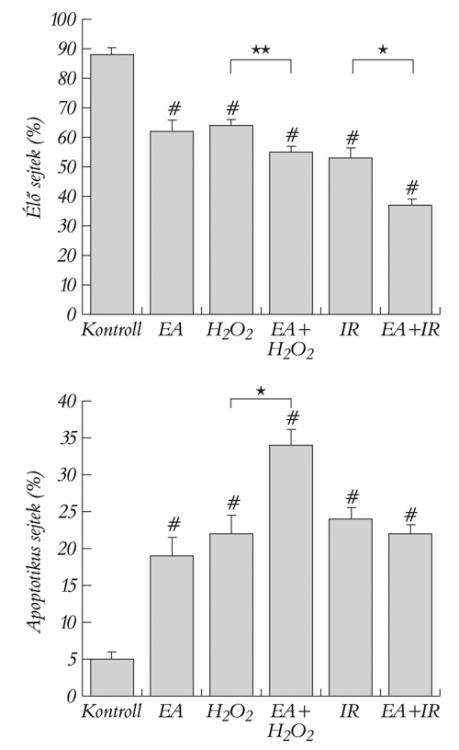
A sejtek életképességét MTT-próbával meghatározva az élő sejtek mennyiségét 100%-nak vettük. Az etakrinsavas gátlás szignifikánsan csökkentette az élő sejtek százalékos arányát (43±11%). A H2O2-vel, előidézett oxidatív stressz és szimulált I/R hasonló mértékű károsodáshoz vezetett. A sejtpusztulás mértéke jelentősen fokozódott, ha a H2O2-vel kezelt csoportok sejtjeihez a GST gátló etakrinsavat is hozzáadtuk.
A sejt apoptózis kialakulását flow-citometriával mértük. A kontrollcsoportban 86±2% volt az élő sejtek (annexin V és PI negatív) és 5±1% volt az apoptózis korai szakaszában lévő sejtek (annexin V pozitív és PI negatív) aránya (1. ábra). EA adását követően, csökkent az élő és növekedett az apoptotikus sejtek százalékos aránya. Az apoptózis mértéke szignifikánsan megemelkedett mind a H2O2, mind a szimulált I/R-kezelt csoportokban, az élő sejtek alacsonyabb számával (1. ábra). Ha a vizsgált médiumhoz még EA-t is adtunk (kettős stresszhatás), az apoptotikus sejtek mennyisége tovább emelkedett, az élő sejtek mennyiségének csökkenésével. Az EA emelte a nekrotikus sejtek számát (annexin V negatív és PI pozitív) szimulált I/R során, ami csökkent élő sejtszámmal járt együtt.
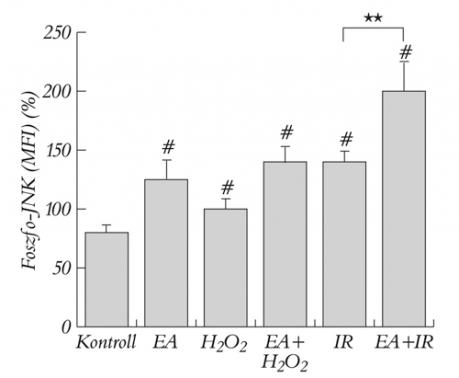
A szívizomsejtekhez adott GST-gátló etakrinsav szignifikánsan emelte a JNK foszforilációját (2. ábra). Amennyiben az oxidatív stresszel egyidejűen GST-gátlást is alkalmaztunk, a JNK aktivációja tovább emelkedett, szignifikáns mértékűvé a szimulált iszkémia-reperfúziós kezelést követően vált (2. ábra).
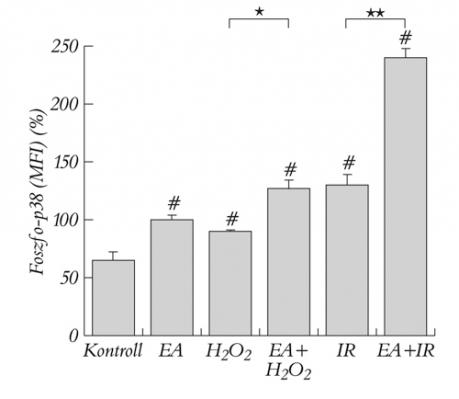
A GST gátlása, a H2O2-vel létrehozott oxidatív stressz és a szimulált iszkémia-reperfúzió a p38 MAPK szignifikáns aktivációjához vezetett a kontrollcsoporthoz képest. Ha a sejteket H2O2 és EA-val együtt inkubáltuk, a foszforilált p38 szintje jelentősen nőtt a csak H2O2-vel kezelt csoporthoz képest. A legkifejezettebb változást a szimulált iszkémia-reperfúzióval együtt alkalmazott GST-gátlás hozta létre, ahol a p38 aktivitása 358±5%-ra emelkedett a kontrollértékekhez képest (3. ábra).
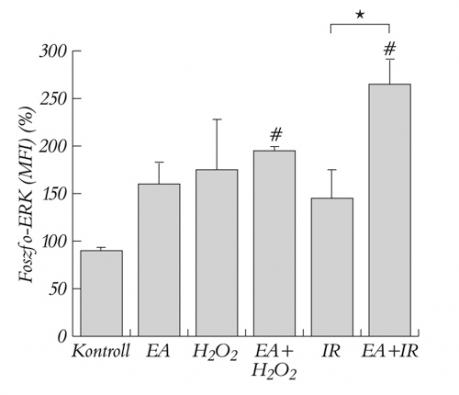
Az ERK/p42-p44 foszforilációja szignifikánsan megemelkedett a GST gátolt csoportokban, ha a sejteket H2O2-dal kezeltük, vagy szimulált I/R-nek tettük ki. A szimulált I/R-nek kitett és az EA-val inkubált szimulált I/R-csoport között szignifikáns eltérés volt kimutatható (4. ábra).
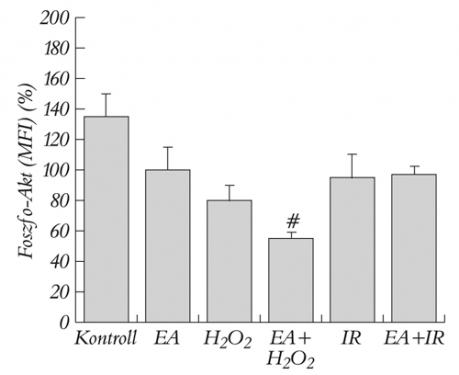
Mind az EA és a H2O2 adása, mind a szimulált I/R-kezelés a PKB/Akt mérsékelt, nem szignifikáns csökkenését okozta. Egyedül a H2O2 kezeléssel együtt alkalmazott GST-gátlás eredményezte a PKB/Akt foszforilációjának szignifikáns csökkenését (5. ábra).
Megbeszélés
Vizsgálataink azt mutatták, hogy a GST farmakológiai gátlása erőteljesen megnöveli az oxidatív stressz indukálta apoptózis mértékét cardiomyocytákban. Ugyanakkor a GST-gátlás a MAP-kinázok fokozott aktivációját okozta szimulált iszkémia-reperfúziót, illetve oxidatív stresszt követően.
Az oxidatív stresszre adott sejtes válaszban a glutation szintje meghatározó és az optimális redukált (GSH) és oxidált (GSSG) arány alapvető a sejt túlélése szempontjából (18, 19). A GST-enzimek feladata a redukált GSH konjugálása idegen vegyületekkel, vagy az oxidatív stressz termékeivel, miáltal kevésbé reaktív anyagok képződnek, amelyek képesek kiürülni a szervezetből (20).
Jelen vizsgálatainkban a GST szerepének tisztázására az enzim etakrinsavval előidézett farmakológiai gátlását használtuk fel. Az EA a GST izoenzimek többségének szubsztrátja. Az etakrinsav GSH-val való nem enzimatikus konjugálása révén keletkező termék (EA-SG) a GST-enzimek gátlószere, sőt nagyobb aktivitással kapcsolódik az enzimhez, mint az EA, ami maga is gátolja a GST-t reverzibilis kovalens kötésen keresztül (21).
EA adása erőteljes emelkedést eredményezett az apoptotikus sejtek számában, főként, ha a sejtek H2O2-vel is kezelve voltak.
Kimutattuk, hogy a GST a MAP-kináz jelátviteli utak szabályozásában fontos szerepet játszik. Irodalmi adatok szerint a GST fehérje-fehérje interakción keresztül endogén inhibitora a c-Jun N-terminális kináznak és ezáltal befolyásolja a stresszre adott sejtes választ és az apoptózist (18). A JNK szerepet játszik az apoptotikus jelátvitelben és citotoxicitást közvetít I/R és oxidatív, nitrozatív stresszel járó különböző állapotokban (22, 23). Stresszmentes állapotokban, a sejtben az alacsony JNK-aktivitást a GST és a JNK által alkotott proteinkomplex tartja fenn (24). Oxidatív, vagy nitrozatív stressz esetén a GST és a JNK disszociál így aktiválódik a JNK, a GST pedig oligomerizáción megy keresztül. A szabaddá vált JNK, foszforilálódik, ezáltal visszanyeri aktivitását, és tovább foszforilálja a c-Jun fehérjét. Hasonló folyamat játszódik le a c-Jun-nal, ami a JNK hatását közvetíti (25). Kísérleteink során kimutattuk, hogy a GST farmakológiai gátlása már önmagában fokozza a JNK aktivitását. Ez magyarázható a JNK GST-vel alkotott fehérje komplex kötés megszüntetésével és az S-glutationiláció gátlásával. Másrészről a GST hatásos gátlása oxidatív károsodást okozhat, a JNK foszforilációjának eredményeként azáltal, hogy stresszmentes állapotban meggátolja a sejtben rendszeresen, kis mennyiségben termelődő oxidánsok és toxikus anyagok eliminációját. Ismert, hogy GST knock-out egerekben magas a JNK alapaktivitása, valamint, hogy sejtek erős GST gátlóval való kezelése a JNK aktivációját okozza (11, 26).
A p38 MAPK-n keresztüli jelátviteli folyamat oxidatív stressz során aktiválódik és befolyásolja a sejtkárosodás mértékét, a stressz válasz közvetítését. Vizsgálataink során azt találtuk, hogy az oxidatív károsodás és szimulált I/R, a p38 aktivitás észrevehető emelkedését okozza szívizomsejtekben, amely tovább növelhető a GST gátlásával.
Eredményeink szerint az ERK/p42-p44 aktiválódott GST-gátlás során, H2O2 adása esetén és reperfúzió alatt. A foszforilált ERK/p42-p44 szintje a szimulált I/R-nek kitett, GST gátolt sejtek esetén meghaladta a foszfo-ERK/p42-p44 szintjét a csak szimulált I/R-rel kezelt sejtekhez képest. Ezek az eredmények az ERK/p42-p44 és a GST közti összefüggést mutatják. Irodalomból ismert, hogy izolált GST p genotípusú immortalizált fibroblaszt szignifikánsan megemelkedett aktivitású ERK/p42-p44-et expresszált (26).
Míg az etakrinsav nem befolyásolta a szimulált I/R-apoptózist fokozó hatását, szignifikánsan nőtt a JNK, p38 és az ERK/p42-p44 foszforilációja. Mivel az EA emelte a nekrotikus sejtek számát és csökkentette az élő sejtszámot mindez arra utal, hogy az etakrinsav jelenlétében szimulált I/R hatására fokozódik a nekrotikus elhalás mértéke.
Kimutatható kapcsolat áll fenn a GST és a PKB/Akt között is. A GST-gátlás hatása a PKB/Akt közvetítette sejtes válaszban nem teljesen tisztázott (27, 28). A PKB/Akt-aktivitás szignifikánsan csökkent azokban a csoportokban, amelyek H2O2 és GST-gátló EA-kezelésben is részesültek, a kontrollértékekhez képest. Az EA-val kezelt sejtek esetén a legátolt antioxidáns, antitoxikus védelem magyarázhatja az általunk kapott eredményeket.
Mindent összevetve, jelen vizsgálataink azt mutatják, hogy a GST gátlása etakrinsavval, súlyosbítja az oxidatív károsodást, és bár a szimulált I/R következtében kialakuló apoptózis mértékét nem fokozza, a nekrotikus sejtpusztulás mértékét tovább növeli. A H2O2-re és szimulált I/R-re a JNK, p38 és ERK/p42-p44 MAPK jelátviteli utak aktiválódtak, amelyet azután az EA szignifikánsan tovább erősített. Eredményeink rámutatnak a GST meghatározó szerepére az oxidatív stressz mértékének kialakításában, és a GST-gátlás további vizsgálatának fontosságára in vivo állatmodellben is.
A kutatásokat támogatta az OTKA- K78434 pályázat.
2. Rőth E, Török B, Zsoldos T, et al. Lipid peroxidation and scavenger mechanism in experimentally induced heart infarcts. Basic Res Cardiol 1985; 80 (5): 530–536.
3. Török B, Rőth E, Matkovics B. Myocardial injuries mediated by free oxygen radicals. Acta Physiol Hung 1986; 68(1): 25–31.
4. Rőth E, Török B, Kelemen D, et al. Free radical mediated injuries after coronary artery occlusion. Basic Res Cardiol 1989; 84 (4): 388–395.
5. Chen QM, Tu VC. Apoptosis and heart failure: mechanisms and therapeutic implications. Am J Cardiovasc Drugs 2002; 2: 43–57.
6. E Rőth, L Hejjel, MT Jaberansari, et al. The role of free radicals in endogenous adaptation and intracellular signals. Exp Clin Cardiol 2004; 9: 13–16.
7. Rőth E, Jaberansari MT. Reactive oxygen species in early and delayed cardiac adaptation. Exp Clin Cardiol 2001; 6(2): 81–6.
8. Sínay L, Kürthy M, Horváth S, et al. Ischaemic postconditioning reduces peroxide formation, cytokine expression and leukocyte activation in reperfusion injury after abdominal aortic surgery in rat model. Clinical Hemorheology and Microcirculation 2008; 40: 133–142.
9. Vinten-Johansen J, Yellon DM, Opie LH. Postconditioning: a simple, clinically applicable procedure to improve revascularization in acute myocardial infarction. Circulation 2005; 4 (112): 2085–2088.
10. Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol 2002; 348: 93–112.
11. Townsend DM, Findlay VL, Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol 2005; 401: 287–307.
12. Wu G, Fang YZ, Yang S, et al. Glutathione metabolism and its implications for health. J Nutr 2004; 134: 489–492.
13. Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother 2003; 57: 145–155.
14. Tokola H, Salo K, Vuolteenaho O, et al. Basal and acidic fibroblast growth factor-induced atrial natriuretic peptide gene expression and secretion is inhibited by staurosporine. Eur J Pharm 1994; 267: 195–206.
15. Luodonpa M, Vuolteenaho O, Eskelinen S, et al. Effects of adrenomedullin on hypertrophic responses induced by angiotensin II, endothelin-1 and phenylephrine. Peptides 2001; 22: 1859–1866.
16. Gordon JM, Dusting GJ, Woodman OL, et al. Cardioprotective action of CRF peptide urocortin against simulated ischemia in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol 2003; 284: 330–336.
17. Vermes I, Haanen C, Steffens-Nakken H, et al. A novel assay for apoptosis. Flow cytometric detection of phospatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods 1995; 184: 39–51.
18. Yin Z, Ivanov VN, Habelhah H, et al. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res 2000; 60: 4053–4057.
19. Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radical Res 1999; 31: 273–300.
20. Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol 2005; 45: 51–88.
21. Tirona RG, Pang KS. Bimolecular glutathione conjugation kinetics of ethacrynic acid in rat liver: in vitro and perfusion studies. J Pharmacol Exp Ther 1999; 290: 1230–1241.
22. Adler V, Yin Z, Fuchs SY et al. Regulation of JNK signaling by GSTp. EMBO J 1999; 18, 1321–1334.
23. Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev 2002; 12: 14–21.
24. Wang T, Arifoglu P, Ronai Z, et al. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem 2001; 276 (24): 20999–21003.
25. Townsend DM. S-glutathionylation: indicator of cell stress and regulator of the unfolded protein response. Mol Interv 2007; 7: 313–24.
26. Ruscoe JE, Rosario LA, Wang T, et al. Pharmacologic or genetic manipulation of glutathione S-transferase P1-1 (GSTpi) influences cell proliferation pathways. J Pharmacol Exp Ther 2001; 298: 339–345.
27. Kim SG, Lee SJ. PI3K, RSK, and mTOR signal networks for the GST gene regulation. Toxicol Sci 2007; 96: 206–213.
28. Kim SK, Novak RF. The role of intracellular signaling in insulin-mediated regulation of drug metabolizing enzyme gene and protein expression. Pharmacol Ther 2007; 113: 88–120.
Cikk értékelése
| Eddig 1 felhasználó értékelte a cikket. |

Hozzászólások